conscious, intentional act or intentional omission of an act that is counter to or violates normal use and is also beyond any further reasonable means of user interface-related risk control by the manufacturer
EXAMPLE: Reckless use or sabotage or intentional disregard of information for safety are such acts.
NOTE 1: See also 4.1.3 of IEC 62366-1:2015.
NOTE 2: An intended but erroneous action that is not abnormal use is considered a type of use error.
NOTE 3: Abnormal use does not relieve the manufacturer from considering non-user interface-related means of risk control.
item, intended specifically by its manufacturer, to be used together with one or more medical devices to specifically enable or assist those medical devices to be used in accordance with their intended use
NOTE 1: An accessory is typically a consumable or separate item for use with one or more medical devices.
NOTE 2: Some authorities having jurisdiction consider an accessory to be a medical device.
NOTE 3: Some authorities having jurisdiction have a different definition of accessory.
an article which, whilst not being itself a medical device, is intended by its manufacturer to be used together with one or several particular medical device(s) to specifically enable the medical device(s) to be used in accordance with its/their intended purpose(s) or to specifically and directly assist the medical functionality of the medical device(s) in terms of its/their intended purpose(s)
an article which, whilst not being itself an in vitro diagnostic medical device, is intended by its manufacturer to be used together with one or several particular in vitro diagnostic medical device(s) to specifically enable the in vitro diagnostic medical device(s) to be used in accordance with its/their intended purpose(s) or to specifically and directly assist the medical functionality of the in vitro diagnostic medical device(s) in terms of its/their intended purpose(s);
materials accompanying a medical device and containing information for the user or those accountable for the installation, use, maintenance, decommissioning and disposal of the medical device, particularly regarding safe use
NOTE 1: The accompanying information shall be regarded as part of the medical device or accessory.
NOTE 2: Accompanying documentation is not necessarily a written or printed document but could involve auditory, visual, or tactile materials and multiple media types.
NOTE 3: Medical devices that can be used safely without instructions for use are exempted from having instructions for use by some authorities with jurisdiction.
information accompanying or marked on a medical device or accessory for the user or those accountable for the installation, use, processing, maintenance, decommissioning and disposal of the medical device or accessory, particularly regarding safe use
NOTE 1: The accompanying information shall be regarded as part of the medical device or accessory.
NOTE 2: The accompanying information can consist of the label, marking, instructions for use, technical description, installation manual, quick reference guide, etc.
NOTE 3: Accompanying information is not necessarily a written or printed document but could involve auditory, visual, or tactile materials and multiple media types (e.g., CD/DVD-ROM, USB stick, website).
NOTE 4: see figure 1 in EN ISO 15223-1:2021
NOTE 5: The label can include the information on the packaging of the medical device.
NOTE 6: e-documentation can include any or all types of information supplied by the manufacturer partially or entirely.
NOTE 7: Marketing information is also known as promotional material.
NOTE 8: There is guidance or rationale related to accompanying information in ISO 20417:2021 Annex A.
means any device, the operation of which depends on a source of energy other than that generated by the human body for that purpose, or by gravity, and which acts by changing the density of or converting that energy. Devices intended to transmit energy, substances or other elements between an active device and the patient, without any significant change, shall not be deemed to be active devices.
Software shall also be deemed to be an active device.
means any untoward medical occurrence, inappropriate patient management decision, unintended disease or injury or any untoward clinical signs, including an abnormal laboratory finding, in subjects, users or other persons, in the context of a performance study, whether or not related to the device for performance study
means any untoward medical occurrence, unintended disease or injury or any untoward clinical signs, including an abnormal laboratory finding, in subjects, users or other persons, in the context of a clinical investigation, whether or not related to the investigational device
notice issued by the organization, subsequent to delivery of the medical device, to provide supplementary information or to advise on action to be taken in the:
NOTE: Issuance of an advisory notice can be required to comply with applicable regulatory requirements.
for the purposes of the definition of nanomaterial, means a collection of weakly bound particles or aggregates where the resulting external surface area is similar to the sum of the surface areas of the individual components
for the purposes of the definition of nanomaterial, means a particle comprising of strongly bound or fused particles
largest amount of a leachable substance that is deemed acceptable on a daily basis, when taken into the body through exposure to a medical device
NOTE: Allowable limits are expressed in dose to the patient for each applicable exposure period. The units used are mass per unit time, e.g. milligrams per day. These doses represent tolerable risks for medical devices under the circumstances of intended use.
means the ability of a device to correctly detect or measure a particular analyte.
The capability of the method to distinguish between two close concentrations of the target marker/analyte.
The ability of an assay to measure in a sample a particular target measurand in the presence of for example other analyte/marker, matrix, interfering substances/organisms or cross-reactive species/agents.
any natural or legal person established within the Union who has received and accepted a written mandate from a manufacturer, located outside the Union, to act on the manufacturer’s behalf in relation to specified tasks with regard to the latter’s obligations under this Regulation
natural or legal person established within a country or jurisdiction who has received a written mandate from the manufacturer to act on his behalf for specified tasks with regard to the latter’s obligations under that country or jurisdiction’s legislation
The Basic UDI-DI is the primary identifier of a device model. It is the DI assigned at the level of the device unit of use. It is the main key for records in the UDI database and is referenced in relevant certificates and EU declarations of conformity.
See lot.
See lot-number.
See lot-number.
positive impact or desirable outcome of the use of a medical device on the health of an individual or a positive impact on patient management or public health
NOTE: Benefits can include positive impact on clinical outcome, the patient’s quality of life, outcomes related to diagnosis, positive impact from diagnostic devices on clinical outcomes, or positive impact on public health.
numerical factor that takes into account the health benefit from use of the medical device(s) containing the leachable substance in question
the analysis of all assessments of benefit and risk of possible relevance for the use of the device for the intended purpose, when used in accordance with the intended purpose given by the manufacturer
ability of a medical device or material to perform with an appropriate host response in a specific application
combination of the probability of harm to health occurring as a result of adverse reactions associated with medical device or material interactions, and the severity of that harm
freedom from unacceptable biological risk in the context of the intended use
means a measurement reference material used in the calibration of a device
value given by the manufacturer to identify a specific medical device or accessory as it relates to its form/fit, function and process (i.e., manufacturing processes requiring differentiation for the end user)
NOTE 1: A catalogue number shall consist of letters or numbers or a combination of these.
NOTE 2: For the purposes of this document, commercial product code should not be confused with the US FDA ‘product code’ or procode classification.
NOTE 3: Synonyms for catalogue number are “reference number” or “reorder number”.
NOTE 4: See figure 2 of ISO 20417:2021.
a marking by which a manufacturer indicates that a device is in conformity with the applicable requirements set out in this Regulation and other applicable Union harmonisation legislation providing for its affixing
reference material, accompanied by a certificate, one or more whose property values are certified by a procedure which establishes its traceability to an accurate realization of the unit in which the property values are expressed, and for which each certified value is accompanied by an uncertainty at a stated level of confidence.
measurement method which has been certified to show appropriate trueness and precision for its intended purpose and has been officially defined as reference method by a competent body.
process of obtaining chemical information, accomplished either by information gathering or by information generation, for example, by literature review or chemical testing
any synthetic or natural substance that is used in a process for manufacturing materials and/or medical devices, including the base material(s), additives (antioxidants, UV stabilizers, color additives, dyes, etc.), and processing aids (solvents, lubricants, antifoaming agents, etc.)
qualitative and quantitative, if applicable, knowledge related to the configuration, composition and production of the medical device and/or its materials of construction, thereby establishing the identities and amounts of constituents present in the materials and device
NOTE 1: See also 5.2.1, 5.2.2, 5.2.3, and Annex B of ISO 10993-18:2018
NOTE 4: Chemical information can be used to establish the hypothetical worst-case release of chemicals from a medical device, predicated on the circumstance that all chemicals present in the device are released from the device under its clinical conditions of use.
devices shall be divided into classes I, IIa, IIb and III, taking into account the intended purpose of the devices and their inherent risks.
devices shall be divided into classes A, B, C and D, taking into account the intended purpose of the devices and their inherent risks.
capable of being read by a person with normal vision
NOTE: There is guidance or rationale for this definition contained in Clause A.2 in ISO 10993-1:2018
the positive impact of a device related to its function, such as that of screening, monitoring, diagnosis or aid to diagnosis of patients, or a positive impact on patient management or public health.
NOTE: It should be recognized that the concept of clinical benefit for in vitro diagnostic medical devices is fundamentally different from that which applies in the case of pharmaceuticals or of therapeutic medical devices, since the benefit of in vitro diagnostic medical devices lies in providing accurate medical information on patients, where appropriate, assessed against medical information obtained through the use of other diagnostic options and technologies, whereas the final clinical outcome for the patient is dependent on further diagnostic and/or therapeutic options which could be available.
the positive impact of a device on the health of an individual, expressed in terms of a meaningful, measurable, patient-relevant clinical outcome(s), including outcome(s) related to diagnosis, or a positive impact on patient management or public health
information concerning safety or performance that is generated from the use of a device and is sourced from the following:
assessment and analysis of clinical data pertaining to a medical device to verify the clinical safety and performance of the device when used as intended by the manufacturer
clinical data and clinical evaluation results pertaining to a device of a sufficient amount and quality to allow a qualified assessment of whether the device is safe and achieves the intended clinical benefit(s), when used as intended by the manufacturer
clinical data and performance evaluation results pertaining to a device of a sufficient amount and quality to allow a qualified assessment of whether the device is safe and achieves the intended clinical benefit(s), when used as intended by the manufacturer
any systematic investigation involving one or more human subjects, undertaken to assess the safety or performance of a device
a document that describes the rationale, objectives, design, methodology, monitoring, statistical considerations, organisation and conduct of a clinical investigation
means the ability of a device, resulting from any direct or indirect medical effects which stem from its technical or functional characteristics, including diagnostic characteristics, to achieve its intended purpose as claimed by the manufacturer, thereby leading to a clinical benefit for patients, when used as intended by the manufacturer
means the ability of a device to yield results that are correlated with a particular clinical condition or a physiological or pathological process or state in accordance with the target population and intended user.
See catalogue number.
See catalogue number.
means a set of technical and/or clinical requirements, other than a standard, that provides a means of complying with the legal obligations applicable to a device, process or system
a device which is essential for the safe and effective use of a corresponding medicinal product to:
(a) identify, before and/or during treatment, patients who are most likely to benefit from the corresponding medicinal product; or
(b) identify, before and/or during treatment, patients likely to be at increased risk of serious adverse reactions as a result of treatment with the corresponding medicinal product
the ability of a device, including software, when used together with one or more other devices in accordance with its intended purpose, to:
(a) perform without losing or compromising the ability to perform as intended, and/or
(b) integrate and/or operate without the need for modification or adaption of any part of the combined devices, and/or
(c) be used together without conflict/interference or adverse reaction
written, electronic or oral communication that alleges deficiencies related to the identity, quality, durability, reliability, usability, safety or performance of a medical device that has been released from the organization’s control or related to a service that affects the performance of such medical devices.
NOTE: The definition of “complaint” differs from the definition given in ISO 9000:2015.
numerical factor that accounts for patient exposure to many medical devices containing the same leachable substance
NOTE: This factor is used to adjust the product of TI and body mass downward.
Configuration is a combination of items of equipment, as specified by the manufacturer, that operate together as a device to achieve an intended purpose. The combination of items may be modified, adjusted or customized to meet specific needs.
Configurations include inter alia:
the process of demonstrating whether the requirements of this Regulation relating to a device have been fulfilled
a body that performs third-party conformity assessment activities including calibration, testing, certification and inspection
means a substance, material or article intended by its manufacturer to be used to verify the performance characteristics of a device
means action taken to eliminate the cause of a potential or actual non-conformity or other undesirable situation
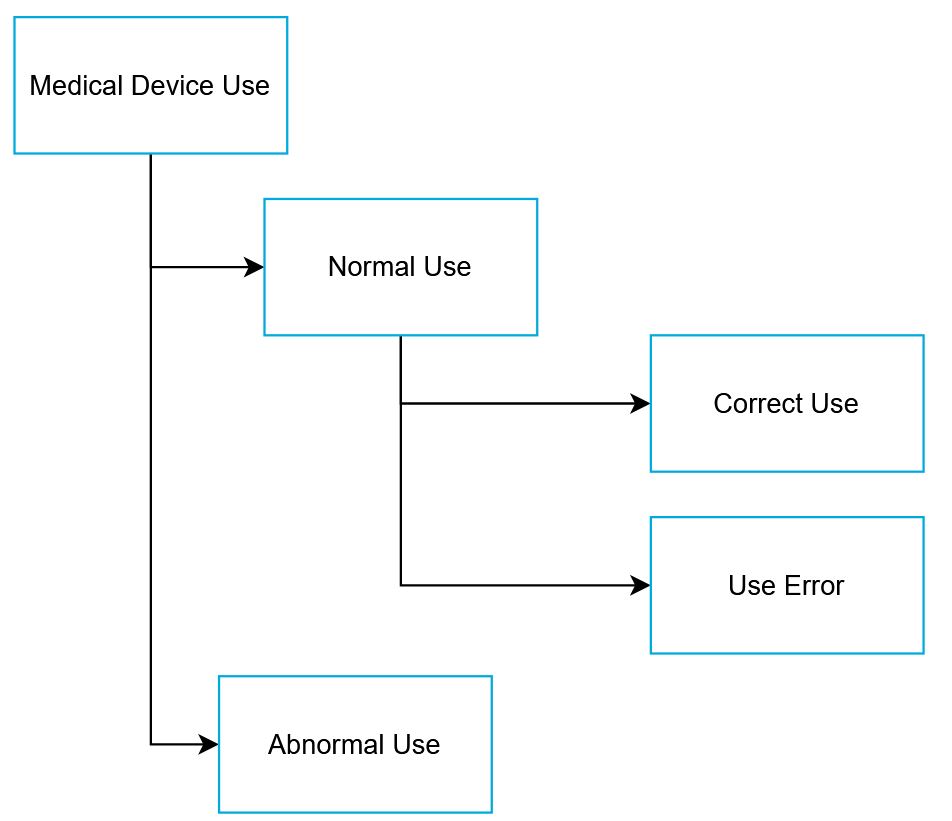
normal use without use error
NOTE 1: Deviation from instructions for use is only considered use error if it leads to a medical device response that is different than intended by the manufacturer or expected by
NOTE 2: See figure under abnormal use, that shows the relationships of the types of use.
any device specifically made in accordance with a written prescription of any person authorised by national law by virtue of that person’s professional qualifications which gives, under that person’s responsibility, specific design characteristics, and is intended for the sole use of a particular patient exclusively to meet their individual conditions and needs
However, mass-produced devices which need to be adapted to meet the specific requirements of any professional user and devices which are mass-produced by means of industrial manufacturing processes in accordance with the written prescriptions of any authorised person shall not be considered to be custom-made devices
information, such as physical and/or chemical characterization, toxicity data, etc. from a variety of sources necessary to characterize the biological response to a medical device
value to be used, in the absence of data, for an uncertainty or other factor used in the calculation of the allowable limit
a ‘non-cellular substance’ extracted from human or animal tissue or cells through a manufacturing process. The final substance used for manufacturing of the device in this case does not contain any cells or tissues
normative text which defines the purpose, the application and the use of the symbol
means any inadequacy in the identity, quality, durability, reliability, safety or performance of a device for performance study, including malfunction, use errors or inadequacy in information supplied by the manufacturer
means any inadequacy in the identity, quality, durability, reliability, safety or performance of an investigational device, including malfunction, use errors or inadequacy in information supplied by the manufacturer
means a device intended by the manufacturer to be used in a performance study.
A device intended to be used for research purposes, without any medical objective, shall not be deemed to be a device for performance study.
any device intended by the manufacturer to be used by lay persons, including devices used for testing services offered to lay persons by means of information society services
any device that is not intended for self-testing but is intended to perform testing outside a laboratory environment, generally near to, or at the side of, the patient by a health professional;
means the ability of a device to identify the presence of a target marker associated with a particular disease or condition.
means the ability of a device to recognise the absence of a target marker associated with a particular disease or condition.
medical device or medical device component that comes into physical contact with body tissue
means any natural or legal person in the supply chain, other than the manufacturer or the importer, that makes a device available on the market, up until the point of putting into service
natural or legal person in the supply chain who, on his own behalf, furthers the availability of a medical device to the end use
NOTE 1: More than one distributor may be involved in the supply chain.
NOTE 2: Persons in the supply chain involved in activities such as storage and transport on behalf of the manufacturer, importer or distributor, are not distributors under this definition.
natural or legal person, different from the manufacturer or importer, in the supply chain who, on their own behalf, furthers the availability of a medical device or accessory to the user
NOTE 1: More than one distributor may be involved in the supply chain.
NOTE 2: For the purposes of this document, persons in the supply chain involved in activities such as storage and transport on behalf of the manufacturer, importer or distributor, are not distributors.
NOTE 3: Distribution activities alone do not include repackaging or otherwise changing the container, wrapper, or accompanying information of the medical device or medical device package other than providing the identification of the distributor.
See single use.
a manufacturer, an authorised representative, an importer or a distributor
a manufacturer, an authorised representative, an importer or a distributor or the person referred to in Article 22(1) and 22(3)
any form of electronically accessible information supplied by the manufacturer related to a medical device or accessory
accuracy and completeness with which users achieve specified goals
NOTE: This is a different concept than ‘clinical effectiveness’.
resources expended in relation to effectiveness
any form of electronically accessible information supplied by the manufacturer related to a medical device or accessory
See clearly legible.
a manufacturer, an authorised representative, an importer, a distributor or the person referred to in MDR Article 22(1) and 22(3)
means an independent body established in a Member State in accordance with the law of that Member State and empowered to give opinions for the purposes of this Regulation, taking into account the views of lay persons, in particular patients or patients’ organisations
European database on medical devices (EUDAMED) is one of the key aspects of the new rules on medical devices (Regulation (EU) 2017/745) and in vitro diagnostic medical devices (Regulation (EU) 2017/746).
EUDAMED will provide a living picture of the lifecycle of medical devices that are made available in the European Union (EU). It will integrate different electronic systems to collate and process information about medical devices and related companies (e.g. manufacturers). In doing so, EUDAMED aims to enhance overall transparency, including through better access to information for the public and healthcare professionals, and to enhance coordination between the different Member States in the EU.
EUDAMED will be composed of six modules related to: actor registration, unique device identification (UDI) and device registration, notified bodies and certificates, clinical investigations and performance studies, vigilance and market surveillance.
time period specified by the manufacturer during which the medical device or accessory is expected to remain safe and effective for use
time period specified by the manufacturer during which the medical device is expected to remain safe for use (i.e. maintain basic safety and essential performance)
NOTE: Maintenance can be necessary during the expected service life.
medical device or medical device component that is partially or wholly located outside the body but has either direct or indirect contact with the internal body fluids and/or tissues
result where the device incorrectly indicates that the specimen tested negative for the condition, attribute or property under analysis
any device with a false presentation of its identity and/or of its source and/or its CE marking certificates or documents relating to CE marking procedures. This definition does not include unintentional non-compliance and is without prejudice to infringements of intellectual property rights
means corrective action taken by a manufacturer for technical or medical reasons to prevent or reduce the risk of a serious incident in relation to a device made available on the market
means a communication sent by a manufacturer to users or customers in relation to a field safety corrective action
medical device or medical device component that has been subjected to all manufacturing processes for the “to be marketed” medical device including packaging and if applicable, sterilization
user interface evaluation conducted with the intent to explore user interface design strengths, weaknesses, and unanticipated use errors
NOTE: Formative evaluation is generally performed iteratively throughout the design and development process, but prior to summative evaluation, to guide user interface design as necessary.
for the purposes of the definition of manufacturer, means the complete rebuilding of a device already placed on the market or put into service, or the making of a new device from used devices, to bring it into conformity with this Regulation, combined with the assignment of a new lifetime to the refurbished device
a set of devices having the same or similar intended purposes or a commonality of technology allowing them to be classified in a generic manner not reflecting specific characteristics
shape and relative arrangement of the parts of the medical device
European standard as defined in point (1)(c) of Article 2 of Regulation (EU) No 1025/2012
potential source of harm
circumstance in which people, property or the environment is/are exposed to one or more hazard
NOTE: See Annex C of ISO 14971:2019 for an explanation of the relationship between hazard and hazardous situation.
Use scenario that could lead to a hazardous situation or harm
NOTE 1: A hazard-related use scenario can often be linked to a potential use error.
NOTE 2: A hazard-related use scenario is not related to a failure of the medical device, unless the medical device failure was caused by a use error.
an organisation the primary purpose of which is the care or treatment of patients or the promotion of public health
any natural or legal person established within the Union that places a device from a third country on the Union market
natural or legal person in the supply chain who is the first in a supply chain to make a medical device, manufactured in another country or jurisdiction, available in the country or jurisdiction where it is to be marketed
natural or legal person who imports a medical device or accessory into a locale that was manufactured in another locale for the purposes of marketing
medical device which is intended to be totally introduced into the human body or to replace an epithelial surface or the surface of the eye by means of clinical intervention and which is intended to remain in place after the procedure
means any device, including those that are partially or wholly absorbed, which is intended:
by clinical intervention and which is intended to remain in place after the procedure. Any device intended to be partially introduced into the human body by clinical intervention and intended to remain in place after the procedure for at least 30 days shall also be deemed to be an implantable device
medical device which can only be removed by medical or surgical intervention and which is intended to:
any medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, piece of equipment, software or system, whether used alone or in combination, intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body, solely or principally for the purpose of providing information on one or more of the following:
(a) concerning a physiological or pathological process or state;
(b) concerning congenital physical or mental impairments;
(c) concerning the predisposition to a medical condition or a disease;
(d) to determine the safety and compatibility with potential recipients;
(e) to predict treatment response or reactions;
(f) to define or monitoring therapeutic measures.
Specimen receptacles shall also be deemed to be in vitro diagnostic medical devices;
device, whether used alone or in combination, intended by the manufacturer for the in vitro examination of specimens derived from the human body solely or principally to provide information for diagnostic, monitoring or compatibility purposes and including reagents, calibrators, control materials, specimen receptacles, software, and related instruments or apparatus or other articles.
means any malfunction or deterioration in the characteristics or performance of a device made available on the market, including use-error due to ergonomic features, as well as any inadequacy in the information supplied by the manufacturer any harm as a consequence of a medical decision, action taken or not taken on the basis of information or result(s) provided by the device
means any malfunction or deterioration in the characteristics or performance of a device made available on the market, including use-error due to ergonomic features, as well as any inadequacy in the information supplied by the manufacturer and any undesirable side-effect
Medical indication is a medical condition which can be treated / diagnosed / prevented / monitored /predicted / prognosed /alleviated.
MEDDEV examples:
This term (medical indication or medical condition) is an umbrella term for all diseases or disabilities that are treated, alleviated , diagnosed etc. by the product. A disease can also be a medical condition.
The term “application area” is another term for Medical indication.
medical device or medical device component through which a fluid or gas passes, prior to the fluid or gas coming into physical contact with body tissue (in this case the medical device or medical device component itself does not physically contact body tissue)
information provided to the user or responsible organization as a risk control measure
EXAMPLE 1: Warnings, precautions or contraindications.
EXAMPLE 2: Instructions for the use of a medical device or accessory to prevent use error or avoid a hazardous situation.
NOTE 1: Information for safety may be found in any or all types of information supplied by the manufacturer.
NOTE 2: Information for safety can be located on the display of a medical device.
information related to the identification and use of a medical device or accessory, in whatever form provided, intended to ensure the safe and effective use of the medical device or accessory
NOTE 1: For the purposes of this document, e-documentation is included in information supplied by the manufacturer.
NOTE 3: The primary purpose of information supplied by the manufacturer is to identify the medical device and its manufacturer, and provide essential information about its safety, performance, and appropriate use to the user or other relevant persons.
NOTE 5: There is guidance or rationale related to information supplied by the manufacturer in Annex A of ISO 20417:2021
means a subject‘s free and voluntary expression of his or her willingness to participate in a particular performance study, after having been informed of all aspects of the performance study that are relevant to the subject’s decision to participate or, in the case of minors and of incapacitated subjects, an authorisation or agreement from their legally designated representative to include them in the performance study
means a subject‘s free and voluntary expression of his or her willingness to participate in a particular clinical investigation, after having been informed of all aspects of the clinical investigation that are relevant to the subject’s decision to participate or, in the case of minors and of incapacitated subjects, an authorisation or agreement from their legally designated representative to include them in the clinical investigation
the information provided by the manufacturer to inform the user of a device’s intended purpose and proper use and of any precautions to be taken
also: package insert
portion of the accompanying information that is essential for the safe and effective use of a medical device or accessory directed to the user of the medical device
NOTE 1: For the purposes of this document, a user can be either a lay user or professional user with relevant specialized training.
NOTE 2: For the purposes of this document, instructions for the professional processing between uses of a medical device or accessory can be included in the instructions for use.
NOTE 3: The instructions for use, or portions thereof, can be located on the display of a medical device or accessory.
NOTE 4: Medical devices or accessories that can be used safely and effectively without instructions for use are exempted from having instructions for use by some authorities having jurisdiction.
NOTE 5: See figure 1 of ISO 20417:2021.
the use for which a device is intended according to the data supplied by the manufacturer on the label, in the instructions for use or in promotional or sales materials or statements and as specified by the manufacturer in the performance evaluation
the use for which a device is intended according to the data supplied by the manufacturer on the label, in the instructions for use or in promotional or sales materials or statements and as specified by the manufacturer in the clinical evaluation
also: intended use
use for which a product, process or service is intended according to the specifications, instructions and information provided by the manufacturer
NOTE: The intended medical indication, patient population, part of the body or type of tissue interacted with, user profile, use environment, and operating principle are typical elements of the intended use.
the ability of two or more devices, including software, from the same manufacturer or from different manufacturers, to:
(a) exchange information and use the information that has been exchanged for the correct execution of a specified function without changing the content of the data, and/or
(b) communicate with each other, and/or
(c) work together as intended
means a clinical performance study where the test results may influence patient management decisions and/or may be used to guide treatment
a device that is assessed in a clinical investigation
means an individual responsible for the conduct of a performance study at a performance study site.
means an individual responsible for the conduct of a clinical investigation at a clinical investigation site
a set of components that are packaged together and intended to be used to perform a specific in vitro diagnostic examination, or a part thereof;
the written, printed or graphic information appearing either on the device itself, or on the packaging of each unit or on the packaging of multiple devices
written, printed, or graphic information appearing on the item (medical device, accessory) itself, on the packaging of each item or on the packaging of multiple items
NOTE 1: For the purposes of document ISO 20417:2021, the term labelled is used to designate the corresponding act.
NOTE 2: Label includes the marking on the medical device or accessory.
NOTE 3: For the purposes of this document, information indicated on a graphical user interface (GUI) is considered as appearing on the item.
NOTE 4: See figure 1 of ISO 20417:2021.
see label
label, instructions for use, and any other information that is related to identification, technical description, intended purpose and proper use of the medical device, but excluding shipping documents
an individual who does not have formal education in a relevant field of healthcare or medical discipline
individual who does not have formal education in a relevant field of healthcare or medical discipline and, if appropriate, relevant specialized training on the use of the specific medical device
NOTE: Additives, sterilant residues, process residues, degradation products, solvents, plasticizers, lubricants, catalysts, stabilizers, anti-oxidants, colouring agents, fillers and monomers, among others.
series of all phases in the life of a medical device, form the initial conception to final decommissioning and disposal
means the likelihood of a given result arising in an individual with the target clinical condition or physiological state compared to the likelihood of the same result arising in an individual without that clinical condition or physiological state
ability to provide measured quantity values that are directly proportional to the value of the measurand in the sample.
An authorised user who has the right to manage certain information regarding the details of the actor and to grant access to Eudamed via the restricted website to other natural persons to act on behalf of that actor;
An authorised user who has the right to grant access to Eudamed via the restricted website to other natural persons to act on behalf of an actor;
also: batch
defined amount of material or a number of medical devices, including finished product and accessories, that is manufactured in one process or a series of related processes and is intended to be homogenous
NOTE 1: A lot or batch is manufactured under essentially the same conditions and is intended to have uniform characteristics and quality within specified limits. A lot or batch is considered homogeneous when equivalent parts or materials are manufactured or tested in the same manner, without interruption, typically on the same day or in the same time period, and produced by the same person or with the same machine/equipment set-up and fulfil the same quality specification.
NOTE 2: The defined amount of material or number of medical devices or accessories is normally associated with a unique statement of conformity to a defined quality specification.
also: batch code, batch number, lot code
production control containing a combination of letters or numbers associated with a single lot or batch
lowest concentration or amount of a substance found by experiment or observation which causes detectable adverse alteration of morphology, functional capacity, growth, development or life span of the target organism under defined conditions of exposure
NOTE: Alterations of morphology, functional capacity, growth, development or life span of the target organism may be detected which are judged not to be adverse.
any supply of a device, other than a device for performance study, for distribution, consumption or use on the Union market in the course of a commercial activity, whether in return for payment or free of charge;
any supply of a device, other than an investigational device, for distribution, consumption or use on the Union market in the course of a commercial activity, whether in return for payment or free of charge
a natural or legal person who manufactures or fully refurbishes a device or has a device designed, manufactured or fully refurbished, and markets that device under its name or trade mark
natural or legal person with responsibility for the design and/or manufacture of a medical device with the intention of making the medical device available for use, under his name, whether or not such a medical device is designed and/or manufactured by that person himself or on his behalf by another person(s).
NOTE 1: The natural or legal person has ultimate legal responsibility for ensuring compliance with all applicable regulatory requirements for the medical device in the countries or jurisdictions where it is intended to be made available or sold, unless this responsibility is specifically imposed on another person by the Regulatory Authority (RA) within that jurisdiction.
NOTE 2: The manufacturer’s responsibilities are described in other GHTF guidance documents. These responsibilities include meeting both pre-market requirements and post-market requirements, such as adverse event reporting and notification of corrective actions.
NOTE 3: “Design and/or manufacture” may include specification development, production, fabrication, assembly, processing, packaging, repackaging, labelling, relabelling, sterilization, installation, or remanufacturing of a medical device; or putting a collection of devices, and possibly other products, together for a medical purpose.
NOTE 4: Any person who assembles or adapts a medical device that has already been supplied by another person for an individual patient, in accordance with the instructions for use, is not the manufacturer, provided the assembly or adaptation does not change the intended use of the medical device.
NOTE 5: Any person who changes the intended use of, or modifies, a medical device without acting on behalf of the original manufacturer and who makes it available for use under his own name, should be considered the manufacturer of the modified medical device.
NOTE 6: An authorised representative, distributor or importer who only adds its own address and contact details to the medical device or the packaging, without covering or changing the existing labelling, is not considered a manufacturer.
NOTE 7: To the extent that an accessory is subject to the regulatory requirements of a medical device, the person responsible for the design and/or manufacture of that accessory is considered to be a manufacturer.
see marking
means the activities carried out and measures taken by competent authorities to check and ensure that devices comply with the requirements set out in the relevant Union harmonisation legislation and do not endanger health, safety or any other aspect of public interest protection
information, in text or graphical format, durably affixed, printed, etched (or equivalent) to a medical device or accessory
NOTE 1: For the purposes of ISO 20417:2021, the term marked is used to designate the corresponding act.
NOTE 2: For the purposes of ISO 20417:2021, marking is different from ‘direct marking’ as commonly described in unique device identification (UDI) standards and regulations. A UDI ‘direct marking’ is a type of marking.
NOTE 3: See Figure 1 of ISO 20417:2021.
synthetic or natural polymer, metal or alloy, ceramic, or composite, including tissue rendered non-viable, used as a medical device or any part thereof
refers to the clinical condition that is to be diagnosed, prevented, monitored, treated, alleviated, compensated for, replaced, modified or controlled by the medical device.
any instrument, apparatus, appliance, software, implant, reagent, material or other article intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the following specific medical purposes:
and which does not achieve its principal intended action by pharmacological, immunological or metabolic means, in or on the human body, but which may be assisted in its function by such means.
The following products shall also be deemed to be medical devices:
instrument, apparatus, implement, machine, appliance, implant, reagent for in vitro use, software, material or other similar or related article, intended by the manufacturer to be used, alone or in combination, for human beings, for one or more of the specific medical purpose(s) of:
and which does not achieve its primary intended action by pharmacological, immunological or metabolic means, in or on the human body, but which may be assisted in its function by such means.
NOTE 1: Products which can be considered to be medical devices in some jurisdictions but not in others include:
group of medical devices manufactured by or for the same organization and having the same basic design and performance characteristics related to safety, intended use and function
see model number
also: model
letters, numbers or a combination of these assigned by a manufacturer to distinguish by function or type, a particular medical device, accessory or medical device family from another
NOTE: See figure 2 of ISO 20417:2021.
medical device or accessory intended by the manufacturer to be reused on multiple patients for multiple uses
NOTE 1: A multiple patient multiple use medical device or accessory typically requires processing between patients.
NOTE 2: A multiple patient multiple use medical device or accessory may require processing between uses on a single patient.
a natural, incidental or manufactured material containing particles in an unbound state or as an aggregate or as an agglomerate and where, for 50 % or more of the particles in the number size distribution, one or more external dimensions is in the size range 1-100 nm;
Fullerenes, graphene flakes and single-wall carbon nanotubes with one or more external dimensions below 1 nm shall also be deemed to be nanomaterials
Material with any external dimension in the nanoscale or having internal structure or surface structure in the nanoscale
means the ability of a device to separate true negative results from false negative results for a given attribute in a given population.
indicates that the medical device or medical device component has neither direct nor indirect contact with body tissues
a conformity assessment body designated in accordance with this Regulation (MDR/IVDR)
having no potential for metabolism or multiplication
operation, including routine inspection and adjustments by any user, and stand-by, according to the instructions for use or in accordance with generally accepted practice for those medical devices provided without instructions for use
NOTE 1: Normal use should not be confused with intended use. While both include the concept of use as intended by the manufacturer, intended use focuses on the medical purpose while normal use incorporates not only the medical purpose, but maintenance, transport, etc. as well.
NOTE 2: Use error can occur in normal use.
NOTE: Objective evidence can be obtained through observation, measurement, test or by other means.
is the packaging in which the device is made available to the end user as intended by the manufacturer.
for the purposes of the definition of nanomaterial, means a minute piece of matter with defined physical boundaries
living being (person) undergoing a medical, surgical or dental procedure
NOTE: A patient can be a user.
means the ability of a device to achieve its intended purpose as claimed by the manufacturer. It consists of the analytical and, where applicable, the clinical performance supporting that intended purpose.
the ability of a device to achieve its intended purpose as stated by the manufacturer
assessment and analysis of data to establish or verify the scientific validity, the analytical and, where applicable, the clinical performance of a device
means a study undertaken to establish or confirm the analytical or clinical performance of a device
means a document that describes the rationale, objectives, design methodology, monitoring, statistical considerations, organisation and conduct of a performance study
See performance.
Manufacturers shall have available within their organisation at least one person responsible for regulatory compliance who possesses the requisite expertise in the field of medical devices.
knowledge regarding formulation, manufacturing processes, geometric and physical properties and type of body contact and clinical use that is used to determine whether any additional biological or material characterization testing is needed
system of modelling biological effects taking into account metabolic and pharmacokinetic differences among species of animal
NOTE: Such data should be utilized whenever they are available.
simplified pictorial representation, used to guide people and tell them how to achieve a certain goal
the first making available of a device, other than a device for performance study, on the Union market;
the first making available of a device, other than an investigational device, on the Union market
means the ability of a device to separate true positive results from false positive results for a given attribute in a given population
means all activities carried out by manufacturers in cooperation with other economic operators to institute and keep up to date a systematic procedure to proactively collect and review experience gained from devices they place on the market, make available on the market or put into service for the purpose of identifying any need to immediately apply any necessary corrective or preventive actions
systematic process to collect and analyse experience gained from medical devices that have been placed on the market
part of the life cycle of the medical device after the design has been completed and the medical device has been manufactured
EXAMPLE: Transportation, storage, installation, product use, maintenance, repair, product changes, decommissioning and disposal.
statement that alerts users to special care or activities necessary for safe and effective use of an IVD medical device or to avoid damage to the IVD medical device that could occur as a result of use, including misuse
NOTE 2: Adapted from U.S. Food and Drug Administration, Guidance on Medical Device Patient Labelling; Final Guidance for Industry and FDA, 19 April 2001
means the probability that a person with a positive device test result has a given condition under investigation, or that a person with a negative device test result does not have a given condition
function that involves user interaction that is related to the safety of the medical device
NOTE 1: Often a primary operating function is interacted with by a series of tasks that can be broken down into a series of user interactions
NOTE 2: The concept of safety includes loss or degradation of performance resulting in an unacceptable risk to the patient, including use error that prevents the user from effectively using the medical device to achieve its intended medical purpose. In IEC 60601-1, this is referred to as ‘essential performance’.
specified way to carry out an activity or a process
NOTE: Procedures can be documented or not.
a combination of products packaged together and placed on the market with the purpose of being used for a specific medical purpose
set of interrelated or interacting activities that use inputs to deliver an intended result
NOTE 1: Whether the “intended result” of a process is called output, product or service depends on the context of the reference.
NOTE 2: Inputs to a process are generally the outputs of other processes and outputs of a process are generally the inputs to other processes.
NOTE 3: Two or more interrelated and interacting processes in series can also be referred to as a process.
activity to prepare a new or used medical device or accessory for its intended use
result of a process
NOTE 1: There are four generic product categories, as follows:
Many products comprise elements belonging to different generic product categories. Whether the product is then called service, software, hardware or processed material depends on the dominant element. For example, the offered product “automobile” consists of hardware (e.g. tyres), processed materials (e.g. fuel, cooling liquid), software (e.g. engine control software, driver’s manual), and service (e.g. operating explanations given by the salesman).
NOTE 2: Service is the result of at least one activity necessarily performed at the interface between the supplier and customer and is generally intangible. Provision of a service can involve, for example, the following:
Software consists of information and is generally intangible and can be in the form of approaches, transactions or procedures. Hardware is generally tangible and its amount is a countable characteristic. Processed materials are generally tangible and their amount is a continuous characteristic. Hardware and processed materials often are referred to as goods.
NOTE 3: This definition of “product” differs from the definition given in ISO 9000:2015.
see catalogue number
see catalogue number
NOTE: The provision of product does not necessarily infer a commercial or financial arrangement.
numerical factor for patient exposure to a leachable substance that accounts for the fact that a medical device is not typically utilized every day during the entire exposure category of interest
NOTE: This factor is used to adjust the product of TI and body mass upwards.
the stage at which a device, other than a device for performance study, has been made available to the final user as being ready for use on the Union market for the first time for its intended purpose;
the stage at which a device, other than an investigational device, has been made available to the final user as being ready for use on the Union market for the first time for its intended purpose
use of a product or system in a way not intended by the manufacturer, but which can result from readily predictable human behaviour.
NOTE 1: Readily predictable human behaviour includes the behaviour of all types of users, e.g. lay and professional users.
NOTE 2: Reasonably foreseeable misuse can be intentional or unintentional.
means any measure aimed at achieving the return of a device that has already been made available to the end user
document stating results achieved or providing evidence of activities performed
NOTE 1: Records can be used, for example, to formalize traceability and to provide evidence of verification, preventive action and corrective action.
NOTE 2: Generally records need not be under revision control.
use of the same device by the same patient more than once without reprocessing
a process carried out on a used device in order to allow its safe reuse including cleaning, disinfection, sterilisation and related procedures, as well as testing and restoring the technical and functional safety of the used device
risk remaining after risk control measures have been implemented
entity accountable for the use and maintenance of a medical device or combination of medical devices
NOTE 1: The accountable entity can be, for example, a hospital, an individual clinician or a lay person. In home use applications, the patient, user and responsible organization can be one and the same person.
NOTE 2: Education and training are included in “use”.
overall process comprising a risk analysis and a risk evaluation
systematic application of management policies, procedures and practices to the tasks of analysing, evaluating, controlling and monitoring risk
set of records and other documents that are produced by risk management
Documented output of the risk management review
Review of the execution of the risk management plan by the manufacturer.
freedom from unacceptable risk
sign giving a general safety message, obtained by a combination of a colour and geometric shape and which, by the addition of a graphical symbol, gives a particular safety message
means the association of an analyte with a clinical condition or a physiological state
production control containing a combination of letters or numbers, selected by the manufacturer, intended for quality control and identification purposes to uniquely distinguish an individual medical device from other medical devices with the same catalogue number or model number
means any adverse event that led to any of the following:
(a) a patient management decision resulting in death or an imminent life-threatening situation for the individual being tested, or in the death of the individual’s offspring,
(b) death,
(c) serious deterioration in the health of the individual being tested or the recipient of tested donations or materials, that resulted in any of the following:
(i) life-threatening illness or injury,
(ii) permanent impairment of a body structure or a body function,
(iii) hospitalisation or prolongation of patient hospitalisation,
(iv) medical or surgical intervention to prevent life-threatening illness or injury or permanent impairment to a body structure or a body function,
(v) chronic disease,
(d) foetal distress, foetal death or a congenital physical or mental impairment or birth defect
means any adverse event that led to any of the following:
(a) death,
(b) serious deterioration in the health of the subject, that resulted in any of the following:
(i) life-threatening illness or injury,
(ii) permanent impairment of a body structure or a body function,
(iii) hospitalisation or prolongation of patient hospitalisation,
(iv) medical or surgical intervention to prevent life-threatening illness or injury or permanent impairment to a body structure or a body function,
(v) chronic disease,
(c) foetal distress, foetal death or a congenital physical or mental impairment or birth defect
any incident that directly or indirectly led, might have led or might lead to any of the following:
(a) the death of a patient, user or other person,
(b) the temporary or permanent serious deterioration of a patient’s, user’s or other person’s state of health,
(c) a serious public health threat
means an event which could result in imminent risk of death, serious deterioration in a person’s state of health, or serious illness, that may require prompt remedial action, and that may cause significant morbidity or mortality in humans, or that is unusual or unexpected for the given place and time
individuals or entity accountable to the responsible organization that install, assemble, maintain or repair a medical device or accessory
measure of the possible consequences of a hazard
period of time until the expiry date during which a medical device or accessory in its original packaging maintains its stability under the conditions specified in the information supplied by the manufacturer
a container in relation to which traceability is controlled by a process specific to logistics systems
medical device or accessory intended by the manufacturer to be reused on an individual patient for multiple uses
NOTE 1: A single patient multiple use medical device or accessory may require processing between uses.
NOTE 2: For an implantable medical device, the duration of a single use is from implanting to explanting the medical device.
also: do not re-use, use only once
medical device, accessory intended by the manufacturer to be used on an individual patient or specimen during a single procedure and then disposed of
NOTE: A single use medical device or accessory is not intended by its manufacturer to be further processed and used again.
a device that is intended to be used on one individual during a single procedure
device, whether of a vacuum-type or not, specifically intended by its manufacturer for the primary containment and preservation of specimens derived from the human body for the purpose of in vitro diagnostic examination
means any individual, company, institution or organisation which takes responsibility for the initiation, for the management and setting up of the financing of the performance study
means any individual, company, institution or organisation which takes responsibility for the initiation, for the management and setting up of the financing of the clinical investigation
concerning medical device or accessory: ability to maintain safety and performance characteristics within the specifications in information supplied by the manufacturer
NOTE 1: Stability applies to:
NOTE 2: Stability of an IVD reagent or measuring system is normally quantified with respect to time:
NOTE: The state of the art embodies what is currently and generally accepted as good practice in technology and medicine. The state of the art does not necessarily imply the most technologically advanced solution. The state of the art described here is sometimes referred to as the “generally acknowledged state of the art”.
free from viable microorganisms
medical device intended to meet the requirements for sterility
NOTE: The requirements for sterility of a medical device can be subject to applicable regulatory requirements or standards.
means an individual who participates in a clinical investigation
means an individual who participates in a performance study and whose specimen(s) undergo in vitro examination by a device for performance study and/or by a device used for control purposes;
user interface evaluation conducted at the end of the user interface development with the intent to obtain objective evidence that the user interface can be used safely
graphical representation appearing on the label and/or associated documentation of a medical device that communicates characteristic information without the need for the supplier or receiver of the information to have knowledge of the language of a particular nation or people
NOTE: The symbol can be an abstract pictorial or a graphical representation, or one that uses familiar objects, including alphanumeric characters (with sufficient justification).
a combination of products, either packaged together or not, which are intended to be inter-connected or combined to achieve a specific medical purpose
NOTE 1: A task description should include the allocation of activities and operational steps between the user and the medical device.
NOTE 2: Tasks should not be described solely in terms of the functions or features provided by the medical device.
portion of the accompanying information directed to the responsible organization and service personnel that is essential for preparation for the first use and safe use, maintenance or repair as well as processing, transport or storage for the expected lifetime of a medical device
NOTE 1: The technical description may be included in the instructions for use.
NOTE 2: See figure 1 of ISO 20417:2021
NOTE: See ISO/TS 21726 for full context.
tolerable contact exposure to a leachable substance resulting from contact with a medical device
NOTE: It is normally expressed in milligrams per square centimetre of body surface.
product of the tolerable intake, the body mass and the utilization factor
NOTE: It is normally expressed in milligrams per day to the patient.
estimate of the average daily intake of a substance over a specified time period, on the basis of body mass, that is considered to be without appreciable harm to health
NOTE: It is normally expressed in milligrams per kilogram of body mass per day. It is derived as a part of the overall establishment of allowable limits for a leachable substance in a medical device.
risk which is accepted in a given context based upon the current values of society
person or group of people who directs and controls a manufacturer at the highest level
capable of causing an adverse biological response
estimate of the average daily intake of a substance over a specified time period, on the basis of body mass, that is considered to be without appreciable harm to health
probability of a specified degree of an adverse reaction occurring in response to a specified level of exposure
limit, such as a tolerable intake (TI), tolerable exposure (TE), allowable limit (AL) value, or Threshold of Toxicological Concern (TTC) below which adverse effects are not expected for relevant biological endpoints
medical device or medical device component that has a very brief duration of contact with body tissue
means to convey the UDI by using automatic identification and data capture (AIDC) and, if applicable, its human readable interpretation (HRI)
NOTE: UDI carriers can include 1D/linear bar code, 2D/Matrix bar code, RFID, etc.
means to convey the UDI by using automatic identification and data capture (AIDC) and, if applicable, its human readable interpretation (HRI)
NOTE: UDI carriers can include 1D/linear bar code, 2D/Matrix bar code, RFID, etc.
characteristic of the user interface that facilitates use and thereby establishes effectiveness, efficiency and user satisfaction in the intended use environment
NOTE: All aspects of usability, including effectiveness, efficiency and user satisfaction, can either increase or decrease safety
application of knowledge about human behaviour, abilities, limitations, and other characteristics to the design of medical devices (including software), systems and tasks to achieve adequate usability
NOTE: Achieving adequate usability can result in acceptable risk related to use.
application of knowledge about human behaviour, abilities, limitations, and other characteristics to the design of and interactions with an IVD medical devices (including software) to achieve adequate usability.
set of records and other documents that are produced by the usability engineering process
method for exploring or evaluating a user interface with intended users within a specified intended use environment
actual conditions and setting in which users interact with the medical device
NOTE: The conditions of use or attributes of the use environment can include hygienic requirements, frequency of use, location, lighting, noise, temperature, mobility, and degree of internationalization.
user action or lack of user action while using the medical device that leads to a different result than that intended by the manufacturer or expected by the user
NOTE 1: Use error includes the inability of the user to complete a task.
NOTE 2: Use errors can result from a mismatch between the characteristics of the user, user interface, task, or use environment.
NOTE 3: Users might be aware or unaware that a use error has occurred.
NOTE 4: An unexpected physiological response of the patient is not by itself considered use error.
NOTE 5: A malfunction of a medical device that causes an unexpected result is not considered a use error.
NOTE 6: See figure under abnormal use, that shows the relationships of the types of use.
see single use
any healthcare professional or lay person who uses a device
NOTE 1: There can be more than one user of a medical device.
NOTE 2: Common users include clinicians, patients, cleaners, maintenance and service personnel.
subset of intended users who are differentiated from other intended users by factors that are likely to influence usability, such as age, culture, expertise or type of interaction with a medical device
means by which the user and the medical device interact
NOTE 1: Accompanying documentation is considered part of the medical device and its user interface.
NOTE 2: User interface includes all the elements of the medical device with which the user interacts including the physical aspects of the medical device as well as visual, auditory, tactile displays and is not limited to a software interface.
NOTE 3: For the purposes of this standard, a system of medical device can be treated as a single user interface.
process by which the manufacturer explores or assesses the user interactions with the user interface
NOTE 1: A user interface evaluation may consist of one or more of the following techniques, amongst others, usability tests, expert reviews, heuristic analyses, design audits or a cognitive walk through.
NOTE 2: User interface evaluation is frequently performed iteratively throughout the design and development process (this is formative evaluation).
NOTE 3: User interface evaluation is a part of the activities involved in verifying and validating the overall medical device design (this is summative evaluation).
user interface or part of a user interface of a medical device previously developed for which adequate records of the usability engineering process according to IEC 62366-1:2015 are not available
collection of specifications that comprehensively and prospectively describe the user interface of a medical device
summary of the mental, physical and demographic traits of an intended user group, as well as any special characteristics, such as occupational skills, job requirements and working conditions, which can have a bearing on design decisions
specific sequence of tasks performed by a specific user in a specific use environment and any resulting response of the medical device
also: application specification
summary of the important characteristics related to the context of use of the medical device
NOTE 1: The intended medical indication, patient population, part of the body or type of tissue interacted with, user profile, use environment, and operating principle are typical elements of the use specification.
NOTE 2: The summary of the medical device use specification is referred to by some authorities having jurisdiction as the ‘statement of intended use’.
NOTE 3: The use specification is an input to determining the intended use of ISO 14971:2007.
numerical factor used to take into account the utilization of the device in terms of frequency of use and utilization in conjunction with other medical devices that can be reasonably anticipated to contain the same leachable substance
confirmation, through the provision of objective evidence, that specified requirements have been fulfilled
NOTE 1: The objective evidence needed for a verification can be the result of an inspection or of other forms of determination such as performing alternative calculations or reviewing documents.
NOTE 2: The activities carried out for verification are sometimes called a qualification process.
NOTE 3: The word “verified” is used to designate the corresponding status.
NOTE 1: The designation of a hazard alert as a warning is reserved for the most significant consequences.
NOTE 2: The distinction between a warning and a precaution is a matter of degree, considering the likelihood and seriousness of the hazard.
NOTE 3: Use includes use errors and reasonably foreseeable misuse. See ISO 14971 and IEC 62366 for discussions of these concepts.
NOTE 4: Adapted from U.S. Food and Drug Administration, Guidance on Medical Device Patient Labelling; Final Guidance for Industry and FDA, 19 April 2001
means any measure aimed at preventing a device in the supply chain from being further made available on the market
Contact us
You are currently viewing a placeholder content from Vimeo. To access the actual content, click the button below. Please note that doing so will share data with third-party providers.
More InformationYou are currently viewing a placeholder content from YouTube. To access the actual content, click the button below. Please note that doing so will share data with third-party providers.
More InformationYou need to load content from reCAPTCHA to submit the form. Please note that doing so will share data with third-party providers.
More InformationYou are currently viewing a placeholder content from Facebook. To access the actual content, click the button below. Please note that doing so will share data with third-party providers.
More InformationYou are currently viewing a placeholder content from Instagram. To access the actual content, click the button below. Please note that doing so will share data with third-party providers.
More InformationYou are currently viewing a placeholder content from X. To access the actual content, click the button below. Please note that doing so will share data with third-party providers.
More Information